The International Consensus Statement on the diagnosis, multidisciplinary management and lifelong care of individuals with achondroplasia was developed with the primary purpose of facilitating the improvement and standardisation of care for children and adults with achondroplasia worldwide, in order to optimise their clinical outcomes and quality of life. The content below is taken from the publication and is presented in a way to help you view the information that may be most relevant to your needs and interests.
The initial meeting of experts was sponsored by BioMarin Pharmaceuticals Inc., who also provided logistic assistance. The International Consensus Statement publication was funded by BioMarin, but developed independently. The publication should be read in its entirety. This website does not provide medical advice and does not replace the need for independent research, medical judgment, or review and analysis of additional resources.
Savarirayan, R. et al. International Consensus Statement on the diagnosis, multidisciplinary management and lifelong care of individuals with achondroplasia. Nat. Rev. Endocrinol. (2021). https://eprintserver2.medengine.com/cop23un012230/
Prof Savarirayan is the lead author of the International Consensus statement. He lead a group of 55 experts and patient representatives from around the world to develop these guidelines with the sole aim of improving and standardising care of children and adults with achondroplasia.
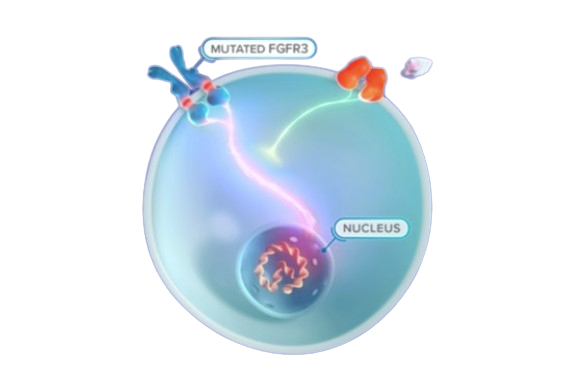
Achondroplasia, the most common skeletal dysplasia, is characterised by a variety of medical, functional and psychosocial challenges across the lifespan. The condition is caused by a common, recurring, gain-of-function mutation in FGFR3, the gene that encodes fibroblast growth factor receptor 3. This mutation leads to impaired endochondral ossification of the human skeleton. The clinical and radiographic hallmarks of achondroplasia make accurate diagnosis possible in most patients. However, marked variability exists in the clinical care pathways and protocols practised by clinicians who manage children and adults with this condition. A group of 55 international experts from 16 countries and 5 continents have developed consensus statements and recommendations that aim to capture the key challenges and optimal management of achondroplasia across each major life stage and sub-specialty area, using a modified Delphi process. The primary purpose of this first International Consensus Statement is to facilitate the improvement and standardisation of care for children and adults with achondroplasia worldwide in order to optimise their clinical outcomes and quality of life.
Achondroplasia is a heritable condition that impairs endochondral ossification, primarily affecting the developing and growing human skeleton (FIG. 1). This condition is the most common skeletal dysplasia with disproportionate short stature, affecting over 360,000 people worldwide1. Important medical, functional and psychosocial challenges exist across the lifespan of individuals with achondroplasia2,3.
The postnatal diagnosis of achondroplasia is fairly straightforward. A combination of key clinical (that is, macrocephaly, short limbed-short stature with rhizomelia and redundant skin folds) and radiographic (that is, characteristic pelvis with short and square ilia, narrow sacro-sciatic notches and narrowing interpedicular distances in the lumbar vertebral spine progressing from L1 to L5) features enables accurate diagnosis in most people with achondroplasia4. Molecular diagnosis is also straightforward because of very limited allelic heterogeneity.
Currently, marked worldwide variability exists in the clinical management of individuals with achondroplasia. This variability leads to variable outcomes with regard to medical, functional and psychosocial consequences of achondroplasia. Furthermore, such differences challenge the ability of health-care professionals, affected individuals and their families, and patient support organisations to objectively evaluate specific management protocols and interventions.
This Consensus Statement is the first to provide international, expert consensus and guidance for the multidisciplinary management of individuals with achondroplasia across their lifespan. We hope that this Consensus Statement might serve as a useful tool to assist in our knowledge about achondroplasia and the management of patients of all ages with achondroplasia.
An initial meeting of 27 participants (comprising 25 health-care professionals and 2 patient representatives from 14 countries, chaired by the first author) was convened in Oslo, Norway, in September 2019 to gain insights into the disease-specific management strategies related to achondroplasia across the lifespan and variations in practice between countries. The meeting was sponsored by BioMarin Pharmaceutical Inc., which provided logistical assistance and had no influence or input regarding the content of this Consensus Statement.
At this meeting, it was first decided that a Consensus Statement to better standardise multidisciplinary management protocols for achondroplasia across the lifespan and including patient perspectives was an unmet global need. Second, it was decided that the group would use a modified Delphi process to capture their combined expertise and experience, based on published evidence whenever possible. Third, the meeting participants decided that other individuals with additional expertise should be invited to participate in the project to ensure global and comprehensive coverage of all aspects of care. Finally, it was decided that the consensus statements derived from this process would be submitted for peer-reviewed publication and dissemination through patient support groups.
Twenty-seven experts in achondroplasia were initially identified and selected based on contributions to the scientific literature and/or on their experience and medical specialty. These experts collectively manage or have managed over 7,000 patients with achondroplasia across all age groups. This initial group selected an additional 28 experts in achondroplasia based on the above criteria to maximise group speciality and geographical representation.
The final International Achondroplasia Consensus Statement Group comprised 55 participants, who are health-care professionals (including 15 medical specialties) and patient representatives, from 16 countries that span 5 continents (Australia, Asia, North America, South America and Europe).
The modified Delphi process began with the face-to-face introductory meeting in Oslo, Norway, that included the initial 27 experts, where these experts selected the broad topics to be developed in the Consensus Statement. Twenty-two sub-teams were then established, drawn from all 55 participants of the International Achondroplasia Consensus Statement Group, to conduct relevant literature reviews and to develop the initial statements. This step was followed by two rounds of voting that included all 55 experts and participants. The participants’ evaluation of each of the statements was captured using a 5-point Likert scale comprising “strongly agree”, “agree”, “neutral”, “disagree”, “strongly disagree” or “abstain”. “Abstain” could be chosen if a participant did not feel they had sufficient clinical knowledge to evaluate a specific statement. The Within3 online platform was used to conduct voting and to collect participant comments and/or feedback on each of the statements (for details of voting, see the Supplementary Dataset).
After the first round of voting by the 55 participants of the International Achondroplasia Consensus Statement Group, 142 statements (of a total of 205) achieved consensus, defined as 80% or more of voting participants indicating “strongly agree” or “agree”; these statements did not undergo a further voting round. Statements that received less than 50% of participants’ votes indicating “strongly agree” or “agree” after the first round of voting were not carried further. Statements that received 50–79% of participants voting “strongly agree” or “agree” were revised by the corresponding sub-teams, guided by the comments and/or feedback collected during the first round; 70 revised statements were submitted for the second round of voting and an additional 18 statements achieved the defined consensus level (Supplementary Dataset). A total of 160 statements achieved consensus after the two-round voting process, comprising this final International Achondroplasia Consensus Statement.
The limitations of this Consensus Statement are chiefly those associated with the underlying Delphi process. Potential confounders of this consensus process include the expert participant selection process and engagement, lack of universal agreement for a ‘consensus’ definition, and the potential for confirmation bias.
Achondroplasia is transmitted in an autosomal dominant manner and is caused by pathogenic missense variants in FGFR3 (often de novo), which encodes fibroblast growth factor receptor 3 (FGFR3). The p.Gly380Arg (c.1138G>A) variant is found in 98–99% of individuals with achondroplasia and p.Gly380Arg (c.1138G>C) is found in 1%. FGFR3 is a tyrosine kinase receptor activated by different fibroblast growth factors and which transmits intracellular signals through the STAT and MAPK pathways5
Although achondroplasia can be diagnosed on a clinical basis without molecular tests, genetic testing can lead to rapid confirmation when limited clinical signs are present (for example, prenatally). Thus, genetic testing can help distinguish achondroplasia from hypochondroplasia or other skeletal dysplasias and can enable increased precision in clinical studies. Skeletal dysplasia gene panels or whole-exome sequencing can be utilised in patients with atypical clinical and/or radiographic presentations.
The International Achondroplasia Consensus Statement Group reached consensus on the following statements relating to the heritability of achondroplasia: 80% of individuals with achondroplasia have unaffected parents who do not have achondroplasia and their condition is caused by a common, de novo, FGFR3 gain-of-function variant. The remaining 20% inherit the causative variant from an affected parent in an autosomal dominant manner. The mutation has 100% penetrance and all individuals who are heterozygous for this mutation will have the clinical manifestations of achondroplasia5. When one parent has achondroplasia, the probability of achondroplasia for each of their future children is 50%. When both parents have achondroplasia, the probability for each of their future children to have average stature is 25%, to have achondroplasia is 50% and to have homozygous achondroplasia (which is usually lethal) is 25%. If one parent has achondroplasia and the other parent has a different but autosomal dominant skeletal dysplasia, the probability of their future children having both achondroplasia and the different skeletal dysplasia is 25%, having solely achondroplasia is 25%, having solely the different skeletal dysplasia is 25%, and having neither achondroplasia nor the different skeletal dysplasia is 25%.
We make the following recommendations for the diagnostics, genetics and molecular testing of achondroplasia.
Recommendation 1. Prenatal genetic testing is possible by chorionic villus sampling or amniocentesis for the FGFR3 pathogenic variant for any pregnancy in which the fetus might have achondroplasia, including ultrasound findings consistent with achondroplasia, if one or both parents has achondroplasia, or if parents of average stature already have a child with achondroplasia, due to the possibility of gonadal mosaicism. Non-invasive prenatal screening using fetal cell-free DNA extracted from maternal blood is available in many but not all locations. Such non-invasive screening might be used if only the father has achondroplasia or if either member of a couple with average stature already had a child with achondroplasia.
Recommendation 2. All adults with achondroplasia should have access to a clinical geneticist or genetic counsellor to discuss inheritance patterns and possibility of prenatal diagnosis, especially in situations where their partner also has a monogenic disorder — ideally, this should be a pre-conception discussion6.
Recommendation 3. When informed about a diagnosis of achondroplasia during the antenatal period, parents should be offered psychological support and be provided information for the management of the child, including future expectations. Introducing parents to advocacy and support groups has been proven beneficial7,8.
Recommendation 4. In infants and young children with suspected achondroplasia, a full skeletal survey is not routinely indicated2,3,9–11.
Recommendation 5. Genetic analysis of the FGFR3 gene can be considered for individuals with clinical and/or radiographic suspicion of achondroplasia to confirm the diagnosis, tailor clinical care and exclude other possible skeletal dysplasias that can mimic achondroplasia.
Recommendation 6. Ultrasound findings of achondroplasia are generally not apparent until 24 weeks of gestation and are often quite subtle5. Genetic testing performed due to the presence of ultrasound anomalies that suggest achondroplasia before 24 weeks of gestation should carefully consider other, more severe, skeletal dysplasias and include testing for these additional skeletal dysplasias unless a parent or sibling has achondroplasia.
Many adults with achondroplasia feel that it is important to have information on the heritability of achondroplasia, the available reproductive options and the potential outcomes for any children that they might have. This information might have been discussed with the parents of the individual when they were young. However, in many cases, the details might have been forgotten or the available diagnostic and treatment options changed in the interim.
We provide the following recommendations for prenatal counselling for adults and families affected by achondroplasia.
Recommendation 7. From adolescence, individuals with achondroplasia or who have partners with achondroplasia should be offered the opportunity to discuss inheritance patterns, pre-conceptual options and any other issues that they might have with healthcare professionals, including geneticists, family counsellors and psychologists. There are several different agencies that would be able to provide this support and information; however, they differ from country to country. It is therefore important for individuals with achondroplasia to identify the most appropriate source of support in each location.
Recommendation 8. People with increased likelihood of conceiving a child with achondroplasia who wish to have prenatal diagnosis in pregnancy should be advised to seek antenatal care as soon as the pregnancy is confirmed; appropriate discussions can then be had with obstetricians and other health-care professionals and arrangements for prenatal diagnosis made.
Recommendation 9. Prenatal diagnosis of achondroplasia is indicated in two situations. The first situation is where there is a family history of achondroplasia, either because of an affected parent or because the child from a previous pregnancy has achondroplasia, and the parents request prenatal diagnosis. The second situation is where there are features detected on fetal ultrasound suggestive of achondroplasia and a definitive diagnosis is required to facilitate accurate prenatal counselling, which should include a discussion of postnatal management and prognosis.
Recommendation 10. When prenatal diagnosis of achondroplasia is indicated, it is recommended that parents meet with specialists experienced in postnatal management of achondroplasia to discuss management and prognosis.
Pregnancy is a life-changing experience for every woman. This International Achondroplasia Consensus Statement Group agrees that pregnancy is manageable for women with achondroplasia6; however, some aspects need to be considered for safety reasons. These include pre-conception medical evaluation, management of pre-term labour and management of the delivery, including location, mode, anaesthesia and perinatal care; caesarean delivery is recommended due to the pelvic anatomy6.
We provide the following recommendations regarding the management of pregnancy in women with achondroplasia.
Recommendation 11. Women with achondroplasia should be offered access to pre-conception evaluation by a health-care provider with an understanding of pregnancy management in women with achondroplasia or other forms of disproportionate short stature and who can assess factors that might impact the safety of pregnancy and birth and optimise pre-conception health6.
Recommendation 12. Women with achondroplasia should receive pregnancy care by an obstetrician, not midwifery-only care. This recommendation is based on the increased need for a caesarean section as well as the potential for respiratory complications of pregnancy6.
Recommendation 13. Pregnant women with achondroplasia should undergo an antenatal anaesthetic clinical assessment, with access to an opinion from an anaesthetist with experience in caring for pregnant women of short stature. There are increased risks of both regional and general anaesthetic in women with achondroplasia. Either can be used for caaesarean delivery, depending on the clinical situation, wishes of the patient and the expert opinion of the anaesthetist6,12.
Recommendation 14. Women with achondroplasia should be assessed on a regular basis throughout their pregnancy for any symptoms suggestive of deterioration in respiratory, cardiac and skeletal status. If there is evidence of compromise, referral for evaluation by the relevant specialist is recommended and there should be access to appropriate investigations (which might include respiratory function tests and echocardiography)6.
Recommendation 15. If either parent has achondroplasia, obstetric ultrasound should be offered during pregnancy every 8 weeks or at a minimum according to the relevant national or local guidelines. This schedule should include at least one ultrasound in the third trimester to maximise the potential of identification of achondroplasia in the fetus6.
Recommendation 16. There should be early discussion between pregnant women with achondroplasia and their antenatal health-care provider about the planned place and mode of birth and anaesthesia options.
Recommendation 17. Women with achondroplasia should be informed that they are not at increased risk of spontaneous preterm birth but that induced preterm birth might be required for maternal reasons6.
Recommendation 18. Management of birth in women with achondroplasia (spontaneous or induced) before fetal viability should be individualised under the care of an experienced obstetrician. Vaginal delivery might be appropriate depending on clinical circumstances6.
Recommendation 19. Delivery beyond 32 weeks should be by caesarean section due to the shape and size of the pelvis in women with achondroplasia. In cases of threatened preterm delivery prior to 32 weeks, an evaluation of fetal size as compared with pelvic size is appropriate, with mode of delivery determined by anticipated cephalopelvic proportion6.
Recommendation 20. The timing of planned birth by caesarean section in women with achondroplasia should be determined by individual clinical circumstances but does not routinely need to be before term6.
Recommendation 21. Planned birth in women with achondroplasia should, when possible, occur in a hospital with on-site critical care facilities and rapid access to blood products and advanced airway equipment6.
Recommendation 22. Given that women with achondroplasia have less than average maternal blood volume, birth attendants should be aware that haemodynamic compromise might occur following birth, even with only moderate blood loss with delivery. Careful fluid management and blood product replacement is essential due to the risk of fluid overload.
Recommendation 23. Postnatal care should, when possible, take place in an environment adapted to allow a woman with achondroplasia to provide usual care for her baby and maximise her recovery, that is, appropriate height beds, alongside cots and accessible toilet facilities.
Awareness and appropriate management of the medical, functional and psychosocial issues that can occur throughout life in people with achondroplasia is key to facilitating optimal outcomes and positively influencing the quality of life of individuals with this condition and their families. Developmental milestones are different for most children with achondroplasia compared with average stature, age-matched peers13,14. During infancy, many have medical complications such as sleep-disordered breathing, otitis media and severe foramen magnum stenosis with cervicomedullary compression, which can increase the risk of sudden death if not diagnosed and treated appropriately3,8,15–17. Conductive hearing loss is common and might affect speech development in children18. Musculoskeletal manifestations, including spinal stenosis and genu varum, are prevalent and can lead to chronic pain in adults with achondroplasia19–21. Physical function and health can decline, notably in the fourth decade of life19,20. Achondroplasia can also be associated with reduced quality of life due to functional impairments and psychosocial challenges and, in some individuals, the condition is associated with reduced life expectancy22–26.
Due to the multisystemic and lifelong effects of achondroplasia, it is critical to adopt a multidisciplinary and pro-active approach to the clinical and psychosocial care of individuals with achondroplasia throughout their life.
Achondroplasia is associated with an increased rate of morbidity and potentially life-threatening medical complications across the lifespan8,16. Many complications, such as sleep-disordered breathing, foramen magnum stenosis and developmental delay, benefit from early intervention and are more common in the first 2 years of life3,16,17. The specialist care team, working in parallel with the local health-care team, should undertake regular evaluation of infants in the first year of life. Frequency should be at least every 2–4 months initially and then every 3–6 months depending on medical concern and local resources8,27.
We provide the following recommendations for the management of achondroplasia in infants.
Recommendation 24. Infants with achondroplasia should be referred to a skeletal dysplasia reference centre or a health-care professional with expertise in achondroplasia as soon as the diagnosis is made.
Recommendation 25. All children with achondroplasia should receive regular follow-up by a multidisciplinary team, guided by a health-care professional with expertise in achondroplasia. Close monitoring in the first 2 years of life is important2,3,15,27.
Recommendation 26. Parents of infants with achondroplasia should be provided with specific charts and a growth parameters register (height, weight and head circumference) for management follow-up28–32.
Recommendation 27. Gross and fine motor developmental milestones are different in infants with achondroplasia as compared with average stature, age-matched peers. Infants with achondroplasia should be assessed for the development of gross, fine motor and early communication skills using achondroplasia-specific screening tools. If developmental delay is observed, MRI of the head and spine and an assessment by a paediatrician and/or neurologist should be considered13,16,27,33.
Recommendation 28. Parents should be provided with early information on positioning and handling infants with achondroplasia, including avoidance of early sitting and appropriate options for car seats and prams8,15,34.
Recommendation 29. Careful evaluation for cervicomedullary compression is mandatory at each medical evaluation in infants and young children with achondroplasia. Signs and symptoms of cervicomedullary compression include motor regression or delayed milestone acquisition, apnoea, difficulty swallowing, poor weight gain, clonus, abnormal reflexes and weakness. Concerning signs or symptoms should be evaluated urgently by a paediatric neurosurgeon3,8,16,35.
Recommendation 30. An increased incidence of sleep-disordered breathing is present in infants with achondroplasia and parents should be informed about typical signs of sleep apnoea. A polysomnography study should be performed when respiratory problems are obvious or suspected but, in any case, completed during the first year of life for all infants with achondroplasia3,8,27,36.
Recommendation 31. Hearing evaluation is recommended in infants with achondroplasia at an early stage and should be monitored longitudinally3,8,27.
Recommendation 32. Recurrent and chronic otitis media are common in infants with achondroplasia and early referral to an otolaryngologist (ear, nose and throat; ENT) specialist should be considered3,8,18,27,36.
Recommendation 33. Infants with achondroplasia should receive regular vaccinations according to national immunisation programmes37.
Foramen magnum stenosis is a well-recognised complication in achondroplasia, with infants and younger children being at higher risk than adults and older children16,17. Many skeletal dysplasia management clinics now screen routinely in the first year with MRI for this complication; if the complication is not present, then high clinical suspicion is still maintained with careful, close monitoring at least up to age 3 years. Clinically, this complication might present with sleep-disordered breathing, hypotonia or hypertonia with increased reflexes and extensor plantar responses; however, children and infants with foramen magnum stenosis can also be asymptomatic. Cervical medullary compression has been implicated as the cause of increased infant death in achondroplasia38,39. It is the consensus of the author panel that symptomatic hydrocephalus is uncommon in achondroplasia40,41.
We make the following recommendations regarding foramen magnum stenosis in infants and children with achondroplasia.
Recommendation 34. Regular neurological evaluation is recommended for all infants and children with achondroplasia3,14,42.
Recommendation 35. MRI is the preferred imaging modality to investigate cervicomedullary compression in infants and children with achondroplasia16,35,43.
Recommendation 36. In asymptomatic infants with achondroplasia, MRI scanning to evaluate the cervicomedullary junction and foramen magnum size should be considered during the first months of life2,14,16,35,43–45.
Recommendation 37. If a cranio-cervical MRI is being performed in infants and children with achondroplasia, imaging of the whole brain should be included40,46.
Recommendation 38. Asymptomatic ventriculomegaly does not require treatment in patients with achondroplasia45,47.
Recommendation 39. An abnormality on neurological evaluation of a patient with achondroplasia, which might indicate cervicomedullary compression, should be evaluated further with MRI16,48.
Recommendation 40. Foramen magnum decompression is indicated in symptomatic children with cervicomedullary compression, with or without MRI signal change in the spinal cord16,42,44,49 (further information can be found in the Respiratory Tract section).
Recommendation 41. Foramen magnum decompression should be carried out by a neurosurgical team with prior experience in performing this procedure in patients with achondroplasia12,46,50.
Recommendation 42. A patient with achondroplasia who has MRI confirmation of cervicomedullary compression, even in the absence of symptoms or signs, should have a neurosurgical evaluation14,16,17,42,49.
Recommendation 43. MRI signal change without concomitant foramen magnum stenosis is not uncommon in the upper cervical cord of older children and adults with achondroplasia and does not usually warrant intervention49,51,52.
Clinical guidelines specify the need to regularly monitor the growth and development of children and adolescents with achondroplasia, including head circumference, height, weight and height-to-weight ratio27. The clinical utility of such assessments is dependent upon the availability of adequate standardised growth charts specific to children and adolescents with achondroplasia29,31,32,53. Due to the anatomical features of children with achondroplasia, developmental milestones differ from those of children without achondroplasia; therefore, condition-specific screening tools should be used. A delay in milestone attainment in comparison to the standard for achondroplasia should be appropriately investigated13.
The effect of growth hormone treatment in children with achondroplasia has been controversial and its long-term effect remains unclear54. The International Achondroplasia Consensus Statement Group reached consensus on the following statements relating to growth hormone treatment. Children with achondroplasia have serum concentrations of growth hormone, insulin-like growth factor 1 and insulin-like growth factor binding protein 1 within normal physiological ranges55. The effects of growth hormone therapy on growth velocity are most likely to be seen in the first 24 months of treatment but the effects on body disproportion are less clear55.
We provide the following recommendations related to growth and development in people with achondroplasia.
Recommendation 44. The growth of infants with achondroplasia should be monitored longitudinally at each medical check-up using achondroplasia-specific height, weight and head circumference growth charts29–32.
Recommendation 45. Head circumference should be monitored regularly (monthly during the first year of life) and plotted on achondroplasia-specific head circumference charts14,27,32. The presence of rapid growth in conjunction with other clinical signs or symptoms of hydrocephalus or cervicomedullary compression might indicate the need for neurosurgical evaluation17.
Recommendation 46. Infants and young children with achondroplasia frequently demonstrate achondroplasia-specific movement strategies that are adaptive for their biomechanical challenges13.
Recommendation 47. Young children with achondroplasia demonstrating delayed development when compared with achondroplasia-specific milestones should be evaluated by medical specialists, physiotherapists, occupational therapists and speech pathologists with skills in this area33,56.
Recommendation 48. Families of infants with achondroplasia should be provided with information and training regarding positioning and handling, including avoidance of early sitting to reduce the potential for development of a fixed thoracolumbar kyphosis and avoiding incidental positional death in car seats2,14,34,57.
Recommendation 49. Young children with achondroplasia typically demonstrate delays in developing independence in certain everyday self-care skills33,56. The need for adaptive equipment, mobility devices or environmental modifications across the home, school, work or community designed to maximise independence should be considered at each medical check-up of individuals with achondroplasia33.
Recommendation 50. Pain is a commonly reported problem across the lifespan in individuals with achondroplasia and should be monitored at each medical check-up19,22.
Recommendation 51. Families and children with achondroplasia should be provided with information regarding the importance of exercise throughout the lifespan in order to maintain strength, healthy body and adipose mass, and flexibility8,14,27,58–60.
Recommendation 52. A DEXA scan is not indicated for bone density assessment in patients with achondroplasia but might be considered to assess body adipose percentage61.
Regular monitoring of children with achondroplasia should be undertaken during clinic visits. Due to the anatomy of children with achondroplasia, gross motor milestones often follow a different pattern to that of children of average stature. Parents should be reassured that this is normal for achondroplasia. It is important to be able to identify when a child falls outside the normal so that appropriate investigations and support can be provided3,8,13.
Children and adults with achondroplasia can find weight loss challenging and obesity is common in this population59,60. Increased weight will have an adverse effect on joints and subsequent mobility and independence. Early encouragement of regular physical activities, such as swimming, coupled with healthy eating will promote a healthy lifestyle, mental wellbeing and prevent obesity62.
Spinal thoracolumbar kyphosis (gibbus) in infants with achondroplasia is common but should resolve when the child begins to mobilise. However, 30% of children with achondroplasia have residual gibbus and associated concerns regarding spinal cord stenosis3. Patients with kyphotic curves between 20–40 degrees a year after mobilisation should be carefully followed and referral to an experienced spinal team should be considered63–66.
Genu varum occurs in children due to ligamental laxity. Children with achondroplasia present continued progression of varus deformity21. Clinical assessment should include asking about activity-induced discomfort or pain3,21.
It is important that therapy advice and medical management should occur in conjunction with a paediatric orthopaedic and/or paediatric spinal surgeon. Surgeons should be familiar with what is normal in achondroplasia and the implications and quality of life effects that spinal or orthopaedic surgery might have on patients with achondroplasia compared with other patients. As such, decisions for operations are best done in the context or in conjunction with the achondroplasia multidisciplinary team.
We make the following recommendations regarding the management of achondroplasia in children.
Recommendation 53. Middle ear effusions are common in children with achondroplasia and can impair hearing. This condition should be screened for with audiology assessments at least annually in early childhood and, if there are concerns, the children should be referred to ENT for consideration of grommets2,3,8,27,36.
Recommendation 54. If there is speech and language delay in the achondroplasia development milestones, then the child should be referred for speech and language therapy36,67.
Recommendation 55. Obstructive sleep apnoea (OSA) can be a common complication of achondroplasia that presents with apnoea or snoring. Parents should be informed of these symptoms and clinicians should ask about OSA during consultations3,8,27,68–70.
Recommendation 56. A healthy lifestyle with emphasis on physical activity and healthy eating should be encouraged during each consultation of a child with achondroplasia.
Recommendation 57. Infants and children with achondroplasia who are noted to be developing behind their peers with achondroplasia when assessed using condition-specific milestone achievement recommendations should be referred to physiotherapists, occupational therapists and speech pathologists with skills in this area13,56.
Recommendation 58. Children with achondroplasia should be reviewed by a physiotherapist and/or occupational therapist with skills in this area to support the development of independence skills, particularly in the area of self-care activities8,15,33.
Recommendation 59. Trips and falls might be common when the child starts to walk. Parents should be encouraged to keep the child active but be aware that they might trip and fall more frequently than children of average stature so appropriate precautions should be taken15.
Recommendation 60. Careful monitoring of the spine should be undertaken in children with achondroplasia. If a kyphosis has not resolved within a year or is progressive in a child who is walking, consultation with a paediatric orthopaedic spine surgeon with experience in achondroplasia is recommended3,63,64,66.
Recommendation 61. Genu varum might start to develop in this age group (age 2–12 years) and become pronounced. If progressive, evaluation by a paediatric orthopaedic surgeon with experience in achondroplasia should be considered2,3,21,71.
Recommendation 62. The possibility of children with achondroplasia undergoing limb lengthening procedures might be discussed and explained to the patient, family and caregivers at this stage. Psychological consultation is advised before undertaking limb lengthening procedures72.
Recommendation 63. During every consultation, the medical team should actively investigate for the presence of pain and/or fatigue in children with achondroplasia. If these symptoms are present, a clinical assessment to determine the cause should be conducted.
Recommendation 64. Surgical interventions should only be performed by surgeons with expertise in achondroplasia and decisions should be made in conjunction with the input of the full achondroplasia multidisciplinary team.
Recommendation 65. Regular dental assessments for all children with achondroplasia should be encouraged and referral to orthodontics should be made when needed73.
Recommendation 66. Children with achondroplasia should use well-fitted car seats for as long as possible and according to local safety standards.
The biomechanical features and disproportionate short stature that are associated with achondroplasia create challenges for individuals with respect to the completion of self-care, driving, independent ambulation and daily functioning. An assessment of function for adolescents and adults with achondroplasia can assist in identifying activity capacity limitations and participation restrictions in this group and direct appropriate and timely service provision or environmental adaptations to assist with independence. Referral of patients to physiotherapists and/or occupational therapists with skills in this area might assist with the timely provision of equipment to maximise independence.
Obesity is a major health problem in achondroplasia necessitating an early yet complex clinical management60,74. Anticipatory care should be directed at identifying children and adolescents who are at high risk of developing obesity. Adolescents with achondroplasia should be provided with information regarding appropriate exercise and/or sports by health practitioners with experience in working with individuals with achondroplasia to minimize the risks of injury or complications. Physical activities will help with mood, mental wellbeing, maintaining an appropriate weight for height, musculoskeletal range and promoting social inclusion62.
Some adolescents might avoid using additional or modified equipment at school due to concerns that it might cause others to tease them or ask questions that they are not comfortable in answering3,26. Their family should be asked if any concerns get in the way of accessing support and then be offered support to help overcome these obstacles.
We present the following recommendations regarding the care of achondroplasia in adolescents.
Recommendation 67. Regular follow-up of adolescents with achondroplasia is recommended, preferably by a medical practitioner or allied health team experienced in the management of this age group8,15,27.
Recommendation 68. The effects that pain has on mood, self-care, education and leisure activities in adolescents with achondroplasia should be specifically evaluated and monitored.
Recommendation 69. Adolescents with symptoms of spinal stenosis should be referred to a spinal service with experience in managing individuals with achondroplasia20,75.
Recommendation 70. The need for adaptive equipment, mobility devices or environmental modifications required in order to maximise independence should be assessed regularly in adolescents with achondroplasia8,15,27.
Recommendation 71. Overweight and obesity issues are common in adolescents with achondroplasia. Monitoring of weight using condition-specific growth and BMI charts and education regarding healthy eating should be provided at each follow-up appointment, involving the wider family.
Recommendation 72. Adolescents with achondroplasia should be encouraged to maintain an active lifestyle62,76,77.
Recommendation 73. Adolescents with achondroplasia might benefit from the support of dedicated professionals to assist with adjustment to their condition and the development of coping strategies for school, employment and social environments8,15.
Although most individuals with achondroplasia lead ordinary lives, the diagnosis might have considerable effects on their physical and mental health20,23,25,26,78. Achondroplasia-related medical complications might occur throughout the entire lifespan, necessitating regular follow-up and management. Symptoms of spinal stenosis and OSA are important to diagnose and manage appropriately20,79–81. Recurrent ear infections in childhood and the craniofacial anatomy in achondroplasia results in a risk of impaired hearing in adulthood82,83. Regular blood pressure monitoring is important, as is preventing weight gain by keeping a healthy diet and regular physical activity84,85. Several studies have reported a high prevalence of pain and declined physical health in adults with achondroplasia that affects daily functioning19,20,22. People with achondroplasia do not seem to be at increased risk of malignancy compared with the rest of the population15,24,38. However, they should participate in appropriate screening programmes (for example, mammography and cervical smear) as recommended for the general population.
The need of relevant helping aids and adaptations at work or at home should be considered to promote optimal participation. Assessment of psychosocial well-being and the potential need for additional support should form part of routine health care for all not just for those with achondroplasia. However, several studies have demonstrated that adults with achondroplasia tend to score lower in quality of life questionnaires for both physical and mental domains than the general population19,22,23,25,26. Appropriate support should be made available to adults with achondroplasia and should be provided in a culturally sensitive way.
We provide the following recommendations for the care of adults with achondroplasia.
Recommendation 74. Ongoing back pain, combined with neurological symptoms, such as claudication, spasticity, reduced walking distance, or bladder and/or bowel dysfunction, might be related to spinal stenosis in adults with achondroplasia and an MRI scan of the entire spine should be considered20,80,86.
Recommendation 75. In the presence of symptomatic spinal stenosis in adults with achondroplasia, where imaging demonstrates evidence of spinal cord impingement or compression, a prompt referral to a spine centre experienced in the management of spinal stenosis in achondroplasia should be considered20,75.
Recommendation 76. In adults with achondroplasia presenting with symptoms suggestive of OSA, an overnight sleep study should be performed81.
Recommendation 77. Blood pressure should be regularly monitored in adults with achondroplasia using a cuff that fits the arm appropriately. Blood pressure measurement on the forearm is an option when elbow contractures or rhizomelia prevents measurement at the upper arm84,87.
Recommendation 78. Adults with achondroplasia might be at increased risk of early-onset hearing loss. There should be a low threshold for assessment of symptomatic individuals and consideration of routine screening at an earlier age than in the general population83,88.
Recommendation 79. Pain should be monitored longitudinally in adults with achondroplasia at each medical check-up. The effect that pain has upon mood, self-care, education, employment and leisure activities in individuals with achondroplasia should be specifically examined and monitored using patient-reported outcome scales such as the Brief Pain Inventory19,20,22,26.
Recommendation 80. Adults with achondroplasia should have routine health checks as advised for the general population in their community.
Recommendation 81. Older adolescents and adults with achondroplasia should be offered genetic counselling to provide information on reproductive options.
Recommendation 82. Adults with achondroplasia should be offered psychosocial support as part of their routine health care, which may include formal assessment by a trained professional23,25.
Recommendation 83. Anaesthesia for people with achondroplasia should be performed by staff experienced with achondroplasia and follow consensus recommendations12.
Recommendation 84. Aids and adaptations, including car adaptations, are required for adults with achondroplasia20.
Access to specialised care is a major challenge in all regions of the world where clinicians with expertise in the management of achondroplasia are unavailable. The consensus statements related to specialty areas therefore focus on the respective key medical issues of concern. They include practical considerations regarding the types and frequency of assessments and interventions in the specialty to provide a global and sustainable approach to care for all individuals with achondroplasia. Relevant literature and evidence have been referenced wherever these exist. The authors consider it paramount that ‘non-expert’ health-care professionals (for example, those with limited or no previous experience in managing patients with achondroplasia) seek expert assistance and guidance or refer their patients to such experts to facilitate the best outcomes.
A thoracolumbar kyphosis is commonly seen in infants with achondroplasia; incidence has been reported to be as high as 90%, decreasing once the child starts mobilising14,34,66,89. Several authors have proposed that development of a fixed thoroacolumbar kyphosis might be exacerbated by prolonged periods of positioning with full spinal flexion2,90. The effects of trunk hypotonia, increased head weight and increased ligamentous laxity combine with the effect of gravity to promote a slumped sitting position, which might contribute to increased anterior wedging of the vertebra and narrowing of the spinal canal34,57,89. In the great majority of patients, the kyphotic deformity resolves spontaneously; however, brace therapy can be considered for children with marked hypotonia and/or motor delay to prevent progression. In situations where fusion and stabilisation of the spine are being considered after decompression spanning more than five spinal levels, the effect of the consequent reduction in lumbar spine mobility on activities of daily living in these patients must be considered, which is further exacerbated by short arms and (often) limited elbow extension75. This author panel agrees that thoracolumbar kyphosis is often seen during infancy in individuals with achondroplasia but resolves without intervention or treatment in the majority of infants when they start to walk34,64,66.
The author panel reached consensus over the following recommendations relating to spine care in individuals with achondroplasia.
Recommendation 85. Thoracolumbar stenosis in individuals with achondroplasia might lead to signs or symptoms of neurogenic claudication. This condition can be mitigated by conservative interventions such as weight loss and physical therapy27. However, if conservative management fails, surgery might be beneficial57,91.
Recommendation 86. Thoracolumbar kyphosis is very common in infants with achondroplasia. All patients should be assessed clinically and, if thoracolumbar kyphosis is pronounced, patients should receive radiographs at baseline and subsequent radiographs as clinically indicated if the kyphosis is progressive3,34,57,64,66,91.
Recommendation 87. At the time of initial surgical decompression of the spine, fusion and stabilisation should be performed in skeletally immature patients with achondroplasia given the propensity for development of post-laminectomy kyphosis due to continued spinal growth63,75.
Recommendation 88. In order to prevent worsening of kyphosis after surgery, fusion and stabilisation are recommended in skeletally mature patients with achondroplasia undergoing spinal decompression spanning more than five levels, crossing a junctional area and in patients with unfavourable sagittal alignment, including thoracolumbar kyphosis75,86.
Recommendation 89. Spinal canal stenosis can lead to signs of myelopathy when it occurs in the cervical and thoracic spine. MRI is recommended for patients with achondroplasia who present with neurological symptoms such as weakness, impaired locomotor and/or fine motor activity, changes in bladder and bowel continence, or pathological reflexes such as hyper-reflexia and clonus20,75,92.
Lower extremity malalignment is frequent in patients with achondroplasia, being complex and multifactorial regarding the aetiology of the deformity21. This deformity is characterised by a three-dimensional, complex and dynamic deformity of the lower limb71 being set apart by genu varum, internal tibial torsion and recurvatum3,93,94. This International Achondroplasia Consensus Statement Group agrees that genu varum is a frequent finding in individuals with achondroplasia and occurs in 40–70% of individuals21. Genu varum can be symmetric or asymmetric and can be associated with or without internal tibial torsion and genu recurvatum3,94. In addition, lower limb malalignment (or genu varum) in individuals with achondroplasia can result in impaired gait and knee or lower leg pain; however, these conditions rarely lead to knee osteoarthritis21,95,96.
We provide the following consensus recommendations for care of genu varum in patients with achondroplasia.
Recommendation 90. The clinical evaluation of lower limb alignment in individuals with achondroplasia should be done in prone, supine and standing positions to analyse internal tibial torsion, mediolateral instability and genu recurvatum as well as during gait to identify lateral thrust3.
Recommendation 91. Assessment of lower limb angular deformity in achondroplasia requires standing anterior-posterior radiographs of the full length of both legs (from hips to ankles) with the patella facing forward71,97–99.
Recommendation 92. Surgical procedures, such as osteotomies or guided growth to correct lower limb malalignment, should be performed in centres of excellence in achondroplasia100. The timing of correction and the technique employed should be tailored to the symptoms, severity, growth patterns and growth remaining3,72.
Recommendation 93. Bracing is not indicated for the treatment of genu varum in patients with achondroplasia95.
Recommendation 94. Surgical indications for correction of lower limb malalignment (or genu varum) in patients with achondroplasia are persistent medial or lateral pain around the knee, instability (lateral thrust), and gait alteration that affects functional and physical capability3,71.
Recommendation 95. In case of lateral knee pain in individuals with achondroplasia, MRI should be considered to rule out the presence of a discoid meniscus, particularly in the absence of angular deformity101–103.
The policies of local advocacy groups regarding limb lengthening have a great influence in the individual decision for practicing this procedure. Although evidence in this area is scarce, limb lengthening is advised in some countries and not recommended in others. Lower and upper limb lengthening should be considered independent procedures depending on the functional, physical and psychosocial requirements of each patient104. The timing of limb lengthening varies and has been performed from early childhood to adult life in individuals with achondroplasia104,105. This Consensus Statement author panel agrees that the indications for limb lengthening for patients with achondroplasia vary widely across countries given differences in personal, cultural, and geographic expectations and opinions. Lower limb lengthening can increase leg length and height, correct malalignment, alter trunk–lower limb proportion, and can improve quality of life104,106. Humeral lengthening has been shown to increase arm length, which alters trunk–upper limb proportion, improves functionality and facilitates self-care since decreased arm span and limited elbow extension are typical in individuals with achondroplasia104,107–109. There is no consensus in the literature on whether limb lengthening surgery should be performed, which surgical methods to use, and the timing to start the first limb lengthening in achondroplasia105.
We provide the following consensus recommendations for limb lengthening in achondroplasia.
Recommendation 96. Specific patient consultation and assessment by a multidisciplinary team should be completed before and after limb lengthening is performed in individuals with achondroplasia to consider and balance all functional, physical and psychosocial outcomes104,110.
Recommendation 97. If a patient chooses to undergo limb lengthening, the procedure should be performed in centres of excellence for patients with achondroplasia. A multidisciplinary team, formed by a paediatric orthopaedic surgeon, anaesthesiologist, physical therapist and paediatrician is necessary in order to achieve better outcomes and prevent complications. Before performing limb lengthening it is necessary to perform whole spine and cervical and skull base MRI to minimise the risk of spinal cord damage caused by neck extension during anaesthesia and surgery.
The majority of individuals with achondroplasia will not develop notable respiratory problems. Chest circumference is small in infants with achondroplasia but catch-up growth in the first 2 years of life usually leads to a normal chest circumference in adults. Nonetheless, respiratory problems should always be considered in the follow-up of patients, especially in combination with gastro-oesophageal reflux.
Individuals with achondroplasia have a high incidence of OSA due to midface hypoplasia111,112 and narrow nares. In infants and young children, central sleep apnoea might be present as well due to spinal cord compression at the cervicomedullary junction16,113. OSA has been reported in 50–80% of children with achondroplasia70,114,115 and in 60% of adults81.
We provide the following consensus recommendations for respiratory issues and sleep-disordered breathing in achondroplasia.
Recommendation 98. Upper airway obstruction and OSA are common in children with achondroplasia and an overnight polysomnography and sleep study should be performed in the first year of life or at the first signs of sleep-disordered breathing, whichever comes earliest; in any case, these studies should be performed no later than at 2 years of age8,12,27,68–70,114–116.
Recommendation 99. The decision to proceed with foramen magnum decompression surgery in achondroplasia is based on clinical and radiological reasons, including the results of polysomnography. The absence of central apnoea on a sleep study should not diminish the indication of a foramen magnum decompression in the presence of clinical and/or MRI criteria. Under those criteria, surgical decompression might lead to neurological and developmental improvement16,115.
Recommendation 100. Tonsillectomy and adenoidectomy is the first-line treatment for OSA in children with achondroplasia68,70,113,114,116,117. Post-operative polysomnography should be completed within 2–4 months after adenotonsillectomy to document improvement or resolution70.
Recommendation 101. Children with achondroplasia undergoing upper airway surgery should be observed for post-operative complications111. Individuals with achondroplasia who undergo surgery to treat OSA should be evaluated for persistent or recurrent disease with follow-up sleep study assessments69,113,117.
Recommendation 102. Additional airway surgeries might be an option for patients with achondroplasia and persistent OSA after adenotonsillectomy. Evaluation by an otolaryngologist or craniofacial surgeon with expertise in achondroplasia is essential to see if additional surgical options exist67,70,115.
Recommendation 103. Children with achondroplasia who have residual OSA following upper airway surgery should be assessed for alternative treatments such as continuous positive airway pressure68,69,118.
Recommendation 104. Individuals with achondroplasia (children or adults) on continuous positive airway pressure for OSA should be assessed by a sleep medicine specialist on a regular basis to determine when repeat sleep studies are required since the severity of OSA can change with time and/or growth.
Recommendation 105. Individuals with achondroplasia are at risk of respiratory failure during respiratory tract infections (for example, bronchiolitis) due to small thoracic volumes and poor pulmonary reserve, justifying preventative measures such as the avoidance of passive smoking and contact with infected persons114. In these patients, respiratory syncytial virus prophylaxis with monoclonal antibodies can be considered.
Recurrent otitis media and hearing loss has been identified in a high number of infants, children and adults with ACH and has been linked to the midface hypoplasia, shortened Eustachian tubes, small pharynx and relative enlargement of tonsils and adenoids18,87,111. A number of teams have recommended that recurrent otitis media in infants and children with ACH should be treated with surgery by placing ear tubes to prevent conductive hearing loss2,36. Hearing loss related to otitis media is thought to be a major contributor to later speech delays and articulation, problems which might have notable effects on future communication, learning and education3.
We provide the following consensus recommendations for ENT care in achondroplasia.
Recommendation 106. Children with achondroplasia should have a comprehensive audiological assessment at birth, as indicated by signs and symptoms or in any case no later than 5 years of age8,27,83.
Recommendation 107. Children with achondroplasia who have speech delay, hearing difficulties, and/or signs and symptoms of middle ear effusion should be referred to an otolaryngologist and undergo a comprehensive audiological assessment8,27,83,111.
Recommendation 108. Adults and children with achondroplasia frequently have hearing loss and middle ear disease. Clinicians should evaluate eardrums and ask about hearing difficulties as part of regular health maintenance to guide appropriate referrals27,83,88.
Recommendation 109. Chronic eustachian tube dysfunction resulting in middle ear effusion and conductive hearing loss is common in patients with achondroplasia. Tympanostomy tube placement can be recommended when middle ear effusions are present for 3 or more months and hearing loss is documented3,18,67,119.
Recommendation 110. Amplification with hearing aids can be offered as a treatment for hearing loss, including patients with conductive hearing loss from middle ear disease67,83.
Recommendation 111. High jugular bulb in the middle ear is more common in patients with achondroplasia; therefore, clinicians should carefully check for otoscopic signs of high jugular bulb before myringotomy or other otological procedures are performed40,120.
During the whole lifespan, preventative dentistry is most important during the establishment of primary dentition. Children with achondroplasia have maxillary hypoplasia, relative mandibular prognathism, class III malocclusion and macroglossia. Teeth have normal number, size and structure but taurodontism of posterior teeth has been reported121. The severity of malocclusion will dictate management. A multidisciplinary check-up of children with achondroplasia should be scheduled at 5–6 years of age, including a specialist in orthodontics together with the breathing specialist and the ENT surgeon.
We provide the following consensus recommendations related to orthodontics and maxillofacial surgery in achondroplasia.
Recommendation 112. In addition to ongoing routine dental care by a general dentist, assessment by a specialist in orthodontics is important in children with achondroplasia given poor maxillary arch growth121.
Recommendation 113. Consider maxillary orthodontic expansion and maxillary non-surgical protraction to increase upper airway volume if, after pharyngeal surgery, there is residual OSA in individuals with achondroplasia115,122.
Recommendation 114. Orthognathic surgical care might be considered after the completion of bone growth in individuals with achondroplasia through Le Fort I or Le Fort III osteotomy. This procedure can be influenced by functional, aesthetic or sleep-related issues118. Clear discussion with the child and family prior to this procedure should occur as facial expression is often altered.
Anaesthesia might be challenging in achondroplasia, especially in older patients. These challenges include difficult intravenous access123 and difficult bag-mask ventilation due to small mouth opening, large tongue, narrow nostrils, midface hypoplasia, large adenoids and short neck with limited head and neck movement12. These difficulties should therefore be planned for.
We provide the following consensus recommendations for anaesthesia in individuals with achondroplasia.
Recommendation 115. Patients with achondroplasia, especially children, should be anaesthetized preferentially in hospitals where care providers are knowledgeable and experienced in caring for patients with achondroplasia and skeletal dysplasias12.
Recommendation 116. Due to foramen magnum stenosis, head and neck movement should be avoided or minimised during bag-mask ventilation and intubation of patients with achondroplasia, especially if the degree of stenosis is unknown, making airway management and intubation more challenging. These difficulties should be planned for by having readily available airway adjuncts for difficult bag-mask ventilation and video laryngoscopes to avoid neck movement12.
Recommendation 117. Thorough pre-assessment is necessary in individuals with achondroplasia and should pay particular attention to airway assessment, range of neck movement, and history of snoring or sleep-disordered breathing113,124. In infants and young children, central sleep apnoea might be present due to cord compression at the cervicomedullary junction. A history of sleep-disordered breathing or results of a sleep study will guide their post anaesthetic stay ranging from same-day discharge, overnight stay, high dependency unit, step-down unit or paediatric intensive care12.
A number of authors have identified a high prevalence of pain in achondroplasia that increases with age and affects daily functioning19,20,22,26. Back pain and neurological symptoms related to spinal stenosis have been identified in a high proportion of adults with achondroplasia20,106.
We provide the following consensus recommendations related to pain and function in individuals with achondroplasia.
Recommendation 118. Pain is a commonly reported problem across the lifespan and should be monitored longitudinally at each medical check-up in individuals with achondroplasia19,22. The effect that pain has upon mood, self-care, education, employment and leisure activities in individuals with achondroplasia should be specifically examined and monitored using patient-reported outcome scales such as the Brief Pain Inventory19.
Recommendation 119. Chronic pain should be considered as a multifactorial problem and is best managed in individuals with achondroplasia through a multidisciplinary approach including neurosurgeons, orthopaedic surgeons, general physicians, pain management specialists, physiotherapists, occupational therapists, psychologists and dieticians15.
Recommendation 120. Ongoing back pain in individuals with achondroplasia, combined with new onset of neurological symptoms, such as claudication, spasticity, reduced walking distance, or bladder or bowel dysfunction, might be related to spinal stenosis and an MRI of the spine should be considered8,20,86,88.
Recommendation 121. When indicated, spinal decompression surgery should be carried out by a team with experience in this procedure in patients with achondroplasia to alleviate pain75.
Recommendation 122. Delayed development of independence for self-care and mobility skills is common in children with achondroplasia compared with average stature children and assessment and management should include a review by an allied health professional, for example, physiotherapists and/or occupational therapists with skills in the assessment and management of children with achondroplasia33.
Recommendation 123. Regular assessment of activities of daily living skills, such as bathing, dressing and toileting, is recommended for children and adults with achondroplasia19,20,33.
Recommendation 124. The need for adaptive equipment, mobility devices or environmental modifications across home, school, work or community, designed to maximise independence are often necessary and therefore individual needs should be considered at each medical check-up for patients with achondroplasia19,20,22.
Individuals with achondroplasia are at disproportionately elevated risk of obesity, which predisposes to breathing difficulties, sleep apnoea, back and joint pain, and reduced mobility60,74,125,126. Current references for diagnosing obesity, estimating metabolic requirements, and informing dietary or physical activity guidelines are not valid when applied to individuals of short and disproportionate stature60,87.
However, at an individual level, anthropometric assessment (that is, height, weight and waist-to-hip ratio) and dietary records should be monitored regularly in adolescents and adults with achondroplasia. Early detection of longitudinal changes in body composition can then be accepted as evidence of energy imbalance (without the need to estimate absolute energy requirements) so that modifiable lifestyle factors such as diet and/or physical activity can be adjusted accordingly. Establishing healthy dietary and physical activity habits early in life is essential and proper early education in relation to diet and nutrition has the potential not only to help individuals enter adult life in a lean condition but also to establish healthy eating patterns that are known to track into adulthood.
We provide the following consensus recommendations relating to nutrition, physical activity and weight management in individuals in achondroplasia.
Recommendation 125. A healthy diet and regular physical activity are recommended for all patients with achondroplasia60,62,85.
Recommendation 126. Modifiable lifestyle factors relevant to obesity risk should be targeted in individuals with achondroplasia to prevent weight gain during childhood and adolescence60.
Recommendation 127. Monitoring of body composition throughout adult life in individuals with achondroplasia is advocated to establish within-patient reference values and detect excess adiposity30,87,125.
More than 80% of individuals born with achondroplasia are part of average height families3 who have no experience in dealing with the diagnosis. The support and positive attitudes of immediate family members as well as friends is a very important social resource for young individuals with achondroplasia and their parents for accepting the diagnosis127,128. This consensus panel agrees that children with achondroplasia can thrive and become adults with independent and fulfilling lives3,8,15,27.
The spectrum of medical complications and the social and psychological burden in children and adults with achondroplasia are well known3,8. Communicating the diagnosis and providing comprehensive guidance to parents and family on the medical, psychological and social aspects related to achondroplasia should be the ultimate goal of health-care professionals. Meetings with other families with a child with achondroplasia are beneficial for most children as they can share experiences and coping strategies. Support and advocacy groups can provide additional support of this type.
We provide the following consensus recommendations related to psychosocial issues, family perspectives and support in achondroplasia.
Recommendation 128. The diagnosis of achondroplasia is associated with varying degrees of psychosocial impact on the individual, the parents and the whole family. Cultural diversity and family dynamics should be taken into consideration to tailor appropriate support for those affected directly or indirectly by achondroplasia129,130.
Recommendation 129. Individuals with achondroplasia can face physical and emotional hurdles across their lifespan and so should be offered or signposted to psychological support when needed. This support can help to develop coping strategies and improve quality of life.
Recommendation 130. Facing social stigma can have a negative emotional impact on the lives of individuals with achondroplasia and for their families. They all should be offered psychological support from health-care professionals and foster emotional resilience and self-esteem together with their peers, friends and advocacy groups to face and overcome different situations.
Recommendation 131. Young individuals with achondroplasia should be offered specific follow-up appointments at the time of transition from paediatric to adult health care in order to facilitate this change in their life and discuss long-term health-care management.
Recommendation 132. Practical, psychosocial and personal needs should be considered and assessed before any changing situation in the educational and professional life of an individual with achondroplasia129,131.
Recommendation 133. Siblings of a child with achondroplasia should be offered informative, psychological and emotional support to help address questions, concerns or conflicts that may arise and highlight their value in the family. Having a child with achondroplasia can be life-changing when the diagnosis is unexpected. Starting from diagnosis, health-care professionals should provide the family with prompt communication, comprehensive guidance and support throughout life3,8,15,27.
Recommendation 134. Understanding the needs and assessing the quality of life of individuals with achondroplasia should be key actions of health-care professionals to assist patients127,128,130,132,133.
Recommendation 135. The medical care of children and adults with achondroplasia should be multidisciplinary. Families should be offered social and psychological support3,7,8,15,27,127,128,130.
Recommendation 136. Awareness and acceptance of achondroplasia can be encouraged and supported through comprehensive health care, advocacy groups and family support. This support will facilitate the building of a positive self-identity, improve wellbeing and increase the quality of life of children and adults with achondroplasia and their families127,128,130,132,133.
Over 360,000 individuals worldwide are estimated to have achondroplasia. This work represents the first global effort to standardise care for individuals with achondroplasia across their lifespan and specialty areas based on available evidence and combined experience. This information will improve clinical management and provide a platform for further research. Key common global challenges and knowledge gaps remain. For example, adequate and equitable access to specialised medical care for individuals with achondroplasia. Furthermore, processes should be developed that allow efficient transitioning of patients from paediatric to adult centres. In addition, detection methods and intervention parameters for foramen magnum and spinal stenosis need to be optimised. Finally, further investigations should be made into the natural history of achondroplasia in adults. Further collaborative research on these issues is a current unmet need. As an increased understanding of the natural history of achondroplasia is gained and as future precision therapies emerge134,135, it will be important to review and revise these guidelines to reflect new knowledge and implement best practices. Additionally, it will be important to observe how the early institution of potential precision therapies for achondroplasia alter its natural history, improve functionality and decrease the need for surgical interventions. However, these therapies will not replace the need for comprehensive, holistic health care, individual and community advocacy, and support of individuals with achondroplasia and their families.
What is the relationship between C-type natriuretic peptide (CNP) and endochondral bone growth?